


















Apert sendromu, doğuştan gelen ve FGFR2 genindeki mutasyonla ortaya çıkan nadir bir genetik bozukluktur. Kafatası kemiklerinin erken kaynaşmasına, yüz ve uzuv şekil bozukluklarına neden olur. Belirgin fiziksel özellikleri arasında yüksek alın, çıkık gözler, orta yüz gelişim geriliği ve sindaktili yer alır. Tanı genellikle doğum sonrası fiziksel bulgularla konur, genetik testle doğrulanır. Tedavi çok sayıda cerrahi müdahale ve destekleyici terapilerle planlanır. Yaşam boyu takip gerektiren sendrom, uygun müdahalelerle normal yaşam süresine olanak tanır.
Apert Sendromu Nedir?
Apert sendromu , bebeklerde kafatası kemiklerinin erken kaynaşmasına neden olan nadir bir genetik hastalıktır. Normalde doğumdan sonra kapanması gereken kafatası kemikleri, bu sendromda gelişim sırasında erken kapanır ve bu durum baş ile yüz şeklinin anormal bir şekilde oluşmasına yol açar.
Doğumsal bir durum olan Apert sendromu, aynı zamanda el ve ayak parmaklarının da birbirine yapışık (sindaktili) olmasına neden olabilir. Bu yapısal bozukluklar, çocuğun hem fiziksel görünümünü hem de bazı organ sistemlerinin işlevlerini etkileyebilir.
Sendroma neden olan genetik mutasyon, kemiklerin birleşme zamanını düzenleyen bir genin yanlış işlemesi sonucu ortaya çıkar. Çoğu vakada bu mutasyon rastlantısal olarak gelişir ve kalıtsal değildir.
Hastalığın kesin bir tedavisi yoktur. Kafatası ve yüz kemiklerine yönelik yapılan cerrahi müdahalelerle çocuğun beyin gelişimi desteklenebilir, solunum gibi yaşamsal fonksiyonlar iyileştirilebilir.
Apert Sendromu Belirtileri
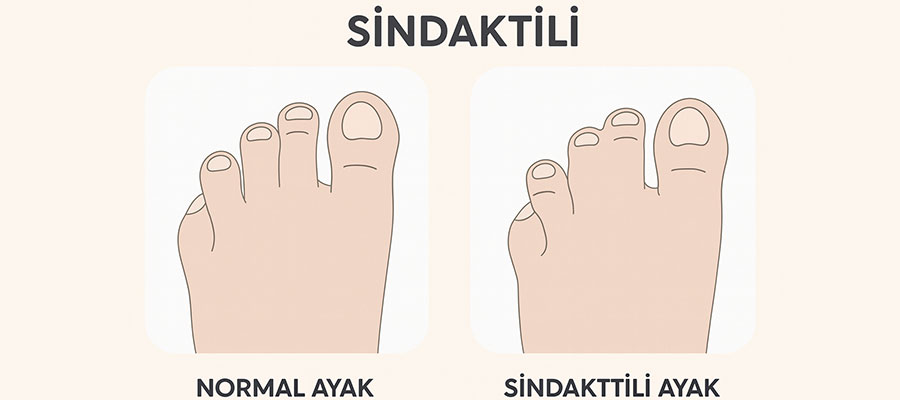
Apert sendromu, doğuştan gelen genetik bir bozukluk olup kafatası, yüz, eller ve ayaklarda belirgin fiziksel farklılıklarla kendini gösterir. Bu sendrom, kemiklerin erken kaynaşması ve parmakların birleşmesi (sindaktili) gibi çok sayıda belirtiye yol açar.
Apert sendromunda sık görülen belirtiler şunlardır:
- Yüksek, geniş ve bombeli alın yapısı
- Başın üst kısmında sivrileşme
- Gözlerin dışa doğru çıkık ve birbirinden uzak olması
- Düz, geniş veya gagamsı burun şekli
- Üst çenenin gelişmemesi ve yüzün orta kısmında çökmüş görünüm
- Yarık damak
- Parmak ve ayak parmaklarının yapışık olması
- Başparmakların kısa ve bükülememesi
- Göz kaslarında dengesizlik nedeniyle şaşılık ve görme problemleri
- Kulak yapısındaki bozukluklara bağlı tekrarlayan kulak enfeksiyonları ve işitme kaybı
- Solunum güçlüğü ve uyku apnesi gibi hava yolu problemleri
- Aşırı terleme (özellikle uykuda) ve yağlı cilt
- Ergenlik döneminde belirginleşen yoğun akne
- Kaş bölgesinde kıl kaybı veya saçsız yamalar
- Hafif veya orta düzeyde zihinsel gelişim geriliği
Apert Sendromu Neden Olur?
Apert sendromu, kemik gelişimini etkileyen genetik bir mutasyon sonucu ortaya çıkan nadir bir hastalıktır. Apert sendromunun temel nedeni, kemik hücrelerinin oluşumunu düzenleyen bir gendeki değişimdir.
Apert sendromunun nedenleri arasında şunlar yer alır:
- FGFR2 geninde mutasyon
- Kemik hücrelerinin erken olgunlaşması
- Erken dönemde kafa kemiklerinin kaynaşması (kraniosinostoz)
- Otosomal dominant kalıtım
- Yeni (de novo) mutasyonlar
- Babaya bağlı ileri yaş etkisi
FGFR2 Geninde Mutasyon
Apert sendromunun nedeni FGFR2 (Fibroblast Büyüme Faktörü Reseptörü 2) geninde meydana gelen mutasyondur. Bu gen, kemik oluşumu ve gelişiminden sorumlu sinyalleri düzenler. Mutasyon sonucu bu sinyaller bozulur ve hücreler kemikleşme sürecine olması gerekenden erken girer. Bu durum, özellikle kafatası ve uzuv kemiklerinde anormal kaynaşmalara yol açar.
Kemik Hücrelerinin Erken Olgunlaşması
FGFR2 genindeki mutasyon, kemik hücrelerinin çok erken bir evrede olgunlaşmasına neden olur. Bu süreçte, kemikleşme henüz tamamlanmadan kemikler birleşir. Büyüme çağındaki çocuğun kafatası ve yüzdeki kemikler genişleyemez ve şekil bozuklukları ortaya çıkar.
Erken Dönemde Kafa Kemiklerinin Kaynaşması (Kraniosinostoz)
Normalde bebeklik döneminde açık olan kafatası sütürleri, Apert sendromunda doğum öncesi dönemde kapanır. Bu duruma kraniosinostoz adı verilir. Kafatası kemiklerinin erken kapanması, beynin büyümesini sınırlar ve basınç artışına, şekil bozukluklarına ve zihinsel gelişim geriliğine neden olabilir.
Otosomal Dominant Kalıtım
Apert sendromu genetik geçişli bir hastalıktır ve otosomal dominant kalıtım yoluyla aktarılabilir. Yani ebeveynlerden biri bu geni taşıyorsa, çocuğuna %50 oranında geçirme ihtimali vardır.
Yeni (De Novo) Mutasyonlar
Vakaların büyük çoğunluğu, ailede daha önce bu hastalık öyküsü olmamasına rağmen ortaya çıkar. Bu tür vakalara de novo (yeni) mutasyonlar denir. Yani mutasyon, doğrudan sperm ya da yumurta hücresinde oluşur ve çocuğa geçer.
Babaya Bağlı İleri Yaş Etkisi
Araştırmalar, ileri yaşta baba olmanın Apert sendromu riskini artırabileceğini göstermektedir. Bu durum, yaşla birlikte sperm hücrelerinde mutasyon gelişme olasılığının artmasından kaynaklanır. Özellikle 40 yaş üstü erkeklerde bu risk daha belirgin hale gelir.
Apert Sendromu Teşhisi Nasıl Konulur?
Apert sendromu teşhisi, genellikle doğumdan kısa süre sonra, yüz ve el-ayak bölgelerinde görülen deformeler fark edildiğinde konulur. Kafatası şekil bozuklukları, yapışık parmaklar ve yüz kemiklerindeki anormallikler, tanı için önemli ipuçlarıdır.
Teşhisi kesinleştirmek amacıyla kranial BT veya MR gibi görüntüleme yöntemleri kullanılırken, FGFR2 gen mutasyonunu saptamak için genetik testler yapılır. Aile öyküsü ve bulgular da tanıya destek sağlar.
Apert Sendromu Tedavisi
Apert sendromunun tedavisi, cerrahi müdahaleler ve tıbbi takipten oluşur. Kafatasındaki erken kemik kaynaşmaları doğumdan sonraki ilk aylarda cerrahiyle düzeltilirken, yüz kemiklerinin ileri alınması ve parmak ayrılması gibi operasyonlar çocukluk döneminde planlanır.
Görme, işitme, solunum ve konuşma gibi fonksiyonel sorunlar destekleyici tedavi ve terapilerle izlenir. Tedavi süreci, plastik cerrahlar, beyin cerrahları, Kulak burun boğaz uzmanları, genetik danışmanlar ve rehabilitasyon ekiplerinin iş birliğiyle yürütülür ve çocuğun gelişimsel ihtiyaçlarına göre kişiselleştirilir.
Kafatası ve Yüz Cerrahisi
Apert sendromunda en erken cerrahi müdahale genellikle 6-8 aylıkken yapılır. Bu dönemde kafatası kemiklerinin erken kaynaşması düzeltilir. Bu işlem, beynin baskı altında kalmasını önler ve normal kafa şeklinin oluşmasını sağlar.
Çocuk büyüdükçe, orta yüz gelişimindeki gerilik belirgin hale gelir. Bu durumda, orta yüz ileri alma cerrahileri uygulanarak hem solunum yolları açılır hem de gözlerin daha iyi korunması sağlanır. Gözler arası mesafenin aşırı geniş olduğu durumlarda ise düzeltme cerrahisi yapılır.
El ve Ayak Cerrahisi
Apert sendromunda ellerdeki sindaktili genellikle ciddi düzeydedir ve çoğu zaman tüm parmaklar birleşmiş durumdadır. Cerrahi ayrılma işlemleri, çocuk yaklaşık 1 yaşına geldiğinde başlar. Ameliyatlarda parmaklar birbirinden ayrılır.
İlerleyen yaşlarda, özellikle 10 yaş civarında, parmakların uzatılması, düzleştirilmesi ya da fonksiyonunun artırılması için ek cerrahiler uygulanabilir. El cerrahisi sonrası her ne kadar tam kavrama gücü sağlanamasa da, çoğu çocuk ellerini kullanabilir.
Solunum, Görme ve İşitme Sorunlarının Yönetimi
Yüz kemiklerindeki deformiteler ve dar burun yapısı, solunum problemlerine yol açabilir. Uyku sırasında hava yolu tıkanıklığı yaşayan çocuklarda CPAP cihazı gibi solunumu destekleyen yöntemler kullanılır. Ciddi durumlarda trakeostomi gerekebilir.
Gözlerin dışa doğru belirgin olması, göz kuruluğu riskini artırır. Göz yüzeyini korumak için gündüz damla, gece ise merhem tedavisi uygulanabilir. Görme problemleri için düzenli göz muayeneleri şarttır.
İşitme kaybı ya da sık tekrarlayan kulak enfeksiyonları olan çocuklarda, kulak tüpü yerleştirilmesi gerekebilir. İşitme cihazları da bu çocuklar için faydalı olabilir.

Apert Sendromu Hakkında Sıkça Sorulan Sorular
Apert sendromu ne kadar yaşar?
Apert sendromlu bireyler uygun cerrahi ve medikal tedaviyle genellikle normal ya da normale yakın bir yaşam süresi yaşayabilir. Özellikle çocukluk dönemindeki ciddi sağlık sorunları kontrol altına alındığında yaşam beklentisi artar.
Apert sendromu özellikleri nelerdir?
Apert sendromu, kafatası şekil bozuklukları, yüz kemiklerinde gerilik, ve el-ayak parmaklarının birleşmesi ile karakterizedir. Görme, işitme, solunum ve zihinsel gelişim gibi alanlarda da etkiler görülebilir.
Apert ve Asperger sendromu nedir?
Apert sendromu, iskelet sistemi ve yüz bölgesindeki bozukluklarla seyreden genetik bir hastalıktır. Asperger sendromu ise otizm spektrum bozuklukları içinde yer alan, sosyal iletişim ve davranışsal zorluklarla tanımlanan nörogelişimsel bir durumdur.
Apert sendromu nasıl kalıtılır?
Apert sendromu otozomal dominant şekilde kalıtılır, yani etkilenen bir ebeveyn hastalığı çocuğuna %50 olasılıkla geçirebilir. Ancak vakaların büyük çoğunluğu yeni mutasyonla oluşur ve ailede hastalık öyküsü olmayabilir.
Apert sendromu genetik mi?
Evet, apert sendromu genetik bir hastalıktır ve FGFR2 genindeki mutasyon sonucu ortaya çıkar. Bu mutasyon, kemik gelişimini kontrol eden sinyal yollarını etkileyerek erken kemik kaynaşmasına yol açar.
Apert sendromu belirtileri nelerdir?
Yüksek alın, çıkık gözler, orta yüz geriliği ve parmakların yapışık olması en sık görülen belirtilerdir. Ayrıca solunum güçlüğü, işitme kaybı, öğrenme zorlukları ve diş problemleri de eşlik edebilir.
Akraba evliliği Apert sendromuna neden olur mu?
Apert sendromu genellikle yeni gen mutasyonlarıyla oluştuğu için akraba evliliğiyle doğrudan ilişkili değildir. Ancak genetik hastalıkların bazı formları akraba evliliklerinde daha sık görülebildiğinden, genetik danışmanlık önerilir.